Current Trends, Challenges, and Optimization Strategies in Bioinformatic Pipelines for Whole Genome Sequencing of Non-Model Species
Keywords:
Whole genome sequencing, Variant calling tools, Pipeline optimization, non-model species, Reference genome bias, Population genomicsAbstract
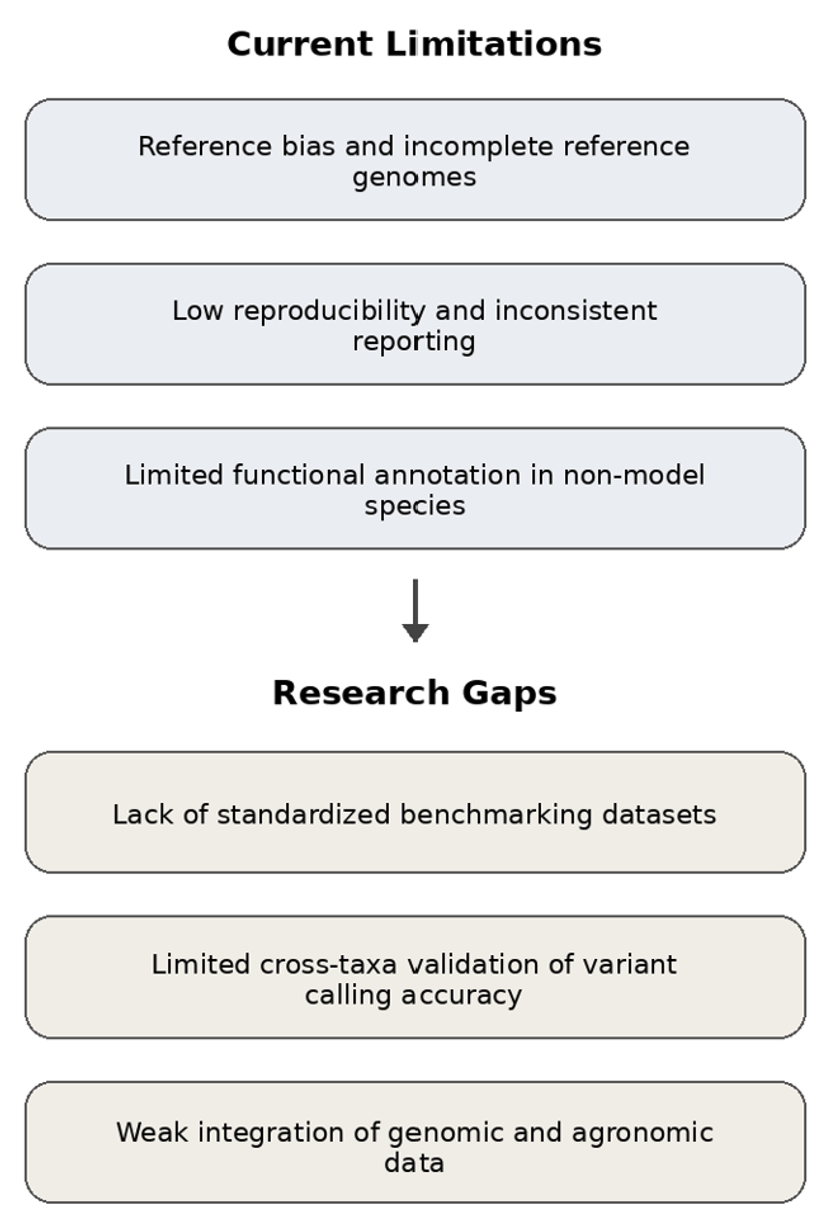
Whole genome sequencing (WGS) has become a central tool in evolutionary biology, conservation genetics, and agricultural genomics, enabling high-resolution analyses of genetic variation across diverse taxa. However, the application of WGS to non-model species presents substantial bioinformatic challenges, including incomplete or biased reference genomes, high levels of genetic diversity, variable sequencing depth, and limited computational resources. These constraints complicate pipeline design, variant discovery, and biological interpretation, particularly in agriculturally relevant systems where genomic outputs must be translated into practical outcomes.
This review critically examines current bioinformatic pipelines used for whole genome sequencing analyses in non-model species, with a focus on methodological trade-offs, sources of bias, and context-dependent optimization strategies. We synthesize recent advances in read processing, alignment and assembly approaches, variant calling frameworks, and functional annotation tools, and compare commonly used pipelines with respect to their suitability for non-model and agricultural applications. In addition, we highlight persistent limitations in benchmarking, reproducibility, and data integration, and discuss emerging trends such as long-read sequencing, pangenome frameworks, and machine learning-assisted pipeline optimization.
By integrating conceptual frameworks, comparative evaluations, and applied examples from crop, livestock, and pathogen genomics, this review provides practical guidance for designing robust and reproducible WGS bioinformatic workflows. The insights presented here aim to support informed methodological decision-making and to facilitate the effective translation of genomic data into agricultural improvement, conservation management, and biological discovery in non-model systems.
References
Bolger, A. M., Lohse, M., & Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, *30*(15), 2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Escalona, M., Rocha, S., & Posada, D. (2016). A comparison of tools for the simulation of genomic next-generation sequencing data. Nature Reviews Genetics, *17*(8), 459–469. https://doi.org/10.1038/nrg.2016.57
Formenti, G., Theissinger, K., Fernandes, C., Bista, I., Bombarely, A., Bleidorn, C., Ciofi, C., Crottini, A., Godoy, J. A., Höglund, J., Malukiewicz, J., Mouton, A., Oomen, R. A., Sadye, P., Palsbøll, P. J., Pampoulie, C., Ruiz-López, M. J., Svardal, H., Theofanopoulou, C., ... & European Reference Genome Atlas (ERGA) Consortium. (2022). The era of reference genomes in conservation genomics. Trends in Ecology & Evolution, *37*(3), 197–202. https://doi.org/10.1016/j.tree.2021.11.005
Garrison, E., & Marth, G. (2012). Haplotype-based variant detection from short-read sequencing. arXiv preprint. arXiv:1207.3907. https://arxiv.org/abs/1207.3907
Günther, T., & Nettelblad, C. (2019). The presence and impact of reference bias on population genomic studies of prehistoric human populations. PLOS Genetics, *15*(7), e1008302. https://doi.org/10.1371/journal.pgen.1008302
Hohenlohe, P. A., Funk, W. C., & Rajora, O. P. (2021). Population genomics for wildlife conservation and management. Molecular Ecology, *30*(1), 62–82. https://doi.org/10.1111/mec.15720
Hoffman, J. I., & Williams, C. L. (2019). A framework for integrating multiple omics datasets to identify genomic features that predict disease risk. Briefings in Bioinformatics, *20*(4), 1301–1312. https://doi.org/10.1093/bib/bbx173
Hogg, C. J., Ottewell, K., Latch, P., Rossetto, M., Biggs, J., Gilbert, A., Godwin, J., Gross, J., Hoeben, P., Holleley, C. E., Hunter, D. A., Lacy, R. C., Lott, M. J., Mastrantonis, S., McDonald, P. G., McLennan, E. A., Peel, E., Pellatt, E. J., Percival-Alwyn, L., ... & Grueber, C. E. (2022). Genomics for conserving threatened species: bridging the gap between theory and practice. Nature Reviews Genetics, *23*(6), 381–393. https://doi.org/10.1038/s41576-022-00458-7
Hotaling, S., Kelley, J. L., & Frandsen, P. B. (2021). Toward a genome sequence for every animal: Where are this section now? Proceedings of the National Academy of Sciences, *118*(52), e2109019118. https://doi.org/10.1073/pnas.2109019118
Huerta-Cepas, J., Szklarczyk, D., Heller, D., Hernández-Plaza, A., Forslund, S. K., Cook, H., Mende, D. R., Letunic, I., Rattei, T., Jensen, L. J., von Mering, C., & Bork, P. (2019). eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Research, *47*(D1), D309–D314. https://doi.org/10.1093/nar/gky1085
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Žídek, A., Potapenko, A., Bridgland, A., Meyer, C., Kohl, S. A. A., Ballard, A. J., Cowie, A., Romera-Paredes, B., Nikolov, S., Jain, R., Adler, J., ... & Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold2. Nature, *596*(7873), 583–589. https://doi.org/10.1038/s41586-021-03819-2
Korneliussen, T. S., Albrechtsen, A., & Nielsen, R. (2014). ANGSD: Analysis of Next Generation Sequencing Data. BMC Bioinformatics, *15*, 356. https://doi.org/10.1186/s12859-014-0356-4
Leprevost, F. da V., Barbosa, V. C., Francisco, E. L., Perez-Riverol, Y., & Carvalho, P. C. (2017). BioContainers: an open-source and community-driven framework for software standardization. Bioinformatics, *33*(16), 2580–2582. https://doi.org/10.1093/bioinformatics/btx192
Leroy, G., Carroll, E. L., Bruford, M. W., DeWoody, J. A., Strand, A., Waits, L., & Wang, J. (2021). Next-generation metrics for monitoring genetic erosion within populations of conservation concern. Evolutionary Applications, *14*(5), 1238–1245. https://doi.org/10.1111/eva.13190
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint. arXiv:1303.3997. https://arxiv.org/abs/1303.3997
Lou, R. N., Jacobs, A., Wilder, A. P., & Therkildsen, N. O. (2021). A beginner’s guide to low‐coverage whole genome sequencing for population genomics. Molecular Ecology, *30*(23), 5966–5993. https://doi.org/10.1111/mec.16077
Mahmoud, M., Gobet, N., Cruz-Dávalos, D. I., Mounier, N., Dessimoz, C., & Sedlazeck, F. J. (2019). Structural variant calling: the long and the short of it. Genome Biology, *20*(1), 246. https://doi.org/10.1186/s13059-019-1828-7
Mölder, F., Jablonski, K. P., Letcher, B., Hall, M. B., Tomkins-Tinch, C. H., Sochat, V., Forster, J., Lee, S., Twardziok, S. O., Kanitz, A., Wilm, A., Holtgrewe, M., Rahmann, S., Nahnsen, S., & Köster, J. (2021). Sustainable data analysis with Snakemake. F1000Research, *10*(33). https://doi.org/10.12688/f1000research.29032.2
O’Neill, M. J., Lawton, B. R., & Rehan, S. M. (2022). Biased representation of genetic variation in non-model species: An evaluation of SNP panels for conservation genomics. Conservation Genetics, *23*(2), 247–260. https://doi.org/10.1007/s10592-021-01415-5
Poplin, R., Ruano-Rubio, V., DePristo, M. A., Fennell, T. J., Carneiro, M. O., Van der Auwera, G. A., Kling, D. E., Gauthier, L. D., Levy-Moonshine, A., Roazen, D., Shakir, K., Thibault, J., Chandran, S., Whelan, C., Lek, M., Gabriel, S., Daly, M. J., Neale, B., MacArthur, D. G., & Banks, E. (2018). Scaling accurate genetic variant discovery to tens of thousands of samples. BioRxiv, 201178. https://doi.org/10.1101/201178
Puritz, J. B., Hollenbeck, C. M., & Gold, J. R. (2014). dDocent: a RADseq, variant-calling pipeline designed for population genomics of non-model organisms. PeerJ, *2*, e431. https://doi.org/10.7717/peerj.431
Reid, J. G., & Lapp, H. (2020). Bioinformatic strategies for analyzing ultra-large-scale sequence data. Current Protocols in Bioinformatics, *70*(1), e102. https://doi.org/10.1002/cpbi.102
Shafer, A. B. A., Wolf, J. B. W., Alves, P. C., Bergström, L., Bruford, M. W., Brännström, I., Colling, G., Dalén, L., De Meester, L., Ekblom, R., Fawcett, K. D., Fior, S., Hajibabaei, M., Hill, J. A., Hoeppner, M. P., Höglund, J., Jensen, E. L., Krause, J., Kristensen, T. N., ... & Zieliński, P. (2017). Genomics and the challenging translation into conservation practice. Trends in Ecology & Evolution, *32*(2), 81–92. https://doi.org/10.1016/j.tree.2016.11.006